1.数据获取
测序数据下载与处理(SRA Toolkit)
测序数据质控与过滤(fastp)
2.序列比对(SAMtools、HISAT2)
3.序列组装(StringTie、TACO)
4.表达定量和差异表达分析(Salmon、DESeq2)
5.GO和KEGG富集分析(clusterProfiler)
☆ RNA-seq方法原理
目的是要给mRNA测序,得到样本的基因表达信息。
llumina的Truseq RNA建库方法:
带Poly(T)探针的磁珠与总RNA进行杂交,吸附其中的带Poly(A)尾巴的mRNA
Mg”离子溶液处理RNA,把RNA打成短片段 被打断的mRNA片段,用随机引物逆转出第一链的cDNA,再合成双链cDNA
在双链CDNA的两端加“A“碱基,并连上”Y“型的接头
经过PCR扩增,成为可以上机的文库
起始总RNA质量控制:用电泳方法。rRNA占有总RNA的大部分,形成的峰越高/尖,RIN(RNA完整度评分值)越高,8以上质量比较好。
测到的RNA片段 mapping到基因组上,进行样品的reads在参考基因上的分布均匀性(Gene coverage)统计。两端平衡的时候表示mRNA降解少(3’高降解多)。
☆ RNA-seq的生物信息分析
一、深度测序数据获取
和EBI、DDBJ组成INSDC,数据内容相同所以找NCBI就行。
(一)NCBI常用数据库
GenBank:遗传序列数据库,收集了所有公开的DNA序列及其注释 GEO (Gene Expression Omnibus)
:收集整理各种表达芯片数据,后来加入了甲基化、lncRNA、miRNA、CNV等其他芯片,还有高通量测序数据。文献中常见GSM和GSE开头的编号,分别是GEO
Sample和GEO Series的数据 PubMed / PMC (PubMed
Central):前者把测序数据和文章联系起来,后者可以进行全文检索,无法访问校园网时可以替代Web of Knowledge
RefSeq:为所有常见生物提供非冗余、人工挑选过的参考序列,通常包含:参考基因组、参考转录组、参考蛋白序列、参考SNP信息、参考CNV信息等等
(二)测序数据的下载和处理:SRA Toolkit
测序数据序列格式
(1)FASTA:表示生物序列的文本格式,基因组和EST序列常常采用
(2)FASTQ格式:表示生物序列及其质量的文本格式
(3)ncbi SRA (Sequence Read Archive) :存储高通量测序原始数据和比对信息,把FASTQ格式文件压缩为SRA格式
绝大多数分析工具不支持SRA,需要使用配套工具包SRA Toolkit先行处理
1. SRA toolkit软件下载
在官网选择适合自己的版本下载。
#我选的ubuntu版本,其他一样,把下载链接修改一下就好了 wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.10.5/sratoolkit.2.10.5-ubuntu64.tar.gz 用conda install sra-tools失败,只好用wget方法或者手动下载到linux盘符下。把安装包下载后用tar xzvf 解压,再配置完PATH就安装好了。
检查配置:
prefetch -V 2.用SRAtoolkit下载并处理NCBI数据
将 .sra文件转换为 .fstaq.gz文件的工具。用NCBI的SRR数据测试一下。
(1)下载
理论上下载东西都可以用wget,但是太慢了。单个数据下载还好,批量下载
prefetch SRRxxxxxxx -O 。
#-O 。 指定到当前路径,否则默认路径难找
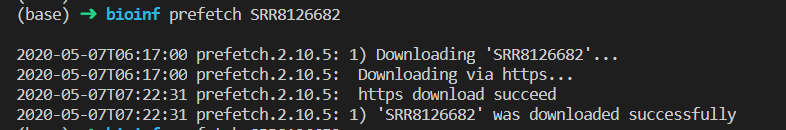
一个数据下了好久,大概1个多小时。不知道怎么优化。
(2)解压
fastq-dump SRRxxxxxxx.sra #解压后从sra文件变为fastq文件
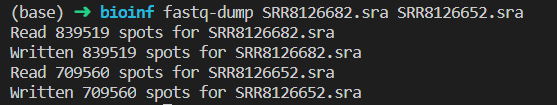
双端测序数据要加–split-files,否则解压后两端的数据不会分开,难以被其他软件读取 如果所用分析软件支持读取gzip,建议加上–gzip,将解压后的数据用gzip压缩,避免占用过多空间
fastq-dump --split-files --gzip xxx.sra
(三)测序数据质控与过滤: fastp
输出HTML和JSON报告,前者方便阅读,后者方便软件读取
单端:fastp -i raw.fq -o clean.fq
双端:fastp -i raw_1.fq -I raw_2.fq -o clean_1.fq -O clean_2.fq
有必要附加的参数:-l 36 -j xxx.json -h xxx.html
默认报告文件名 fastp.json 和 fastp.html,处理多个样本时极易互相覆盖,建议改为样本名称
fastp参数设置
# I/O options 输入输出序列文件 -i 《单端-输入文件名》 -o 《单端-输出文件名》 -I 《双端-输入文件名》 -O 《双端-输出文件名》
#过滤后的最短序列长度 -l 36 #默认15,建议设为36或40 # reporting options 报告参数 -j 《the json format report file name 》 -h 《the html format report file name 》 -R “report_title”
二、序列比对:HISAT2
注释格式介绍
(1)GFF/GTF格式:一般用于基因组和基因注释
(2)SAM格式:储存序列比对到基因组上的信息的文本格式,
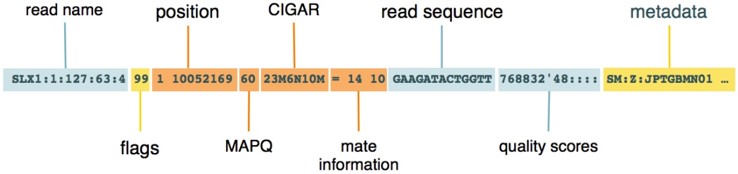
(3)BAMS:SAM的基础上,用二进制 (Binary) 编码,以便压缩体积。
压缩率高于gzip,绝大多数下游分析工具使用
(4)CRAM:在BAM的基础上,借助参考序列,进一步减少空间占用
用SAMtools将SAM转化为BAM或CRAM格式
samtools sort -o xxx.bam xxx.sam samtools sort -o xxx.cram --reference ref.fa -O cram xxx.sam #加-O指定输出格式 建立索引以便快速读取
samtools index xxx.bam samtools index xxx.cram
为什么要比对 (align / map)
locate:测序所得的短序列在基因组的哪个位置
variant:如果个别碱基与基因组不一致,是测序错误还是变异
比对软件工作过程
根据基因组序列FASTA和注释GTF,通过一定的算法编制索引
FASTQ比对到索引,生成SAM文件
如HISAT 和 Bowtie 基于BWT算法。
1. 用HISAT2建立索引
有注释:基因组GTF文件Splice Sites和Exons信息,与基因组序列一起用于建立索引
hisat2_extract_splice_sites.py genes.gtf 》 splicesites.txt hisat2_extract_exons.py genes.gtf 》 exons.txt hisat2-build --ss splicesites.txt --exon exons.txt genome.fa genome 没注释:直接用基因组序列建立索引
hisat2-build genome.fa genome 结果产生索引文件genome(指向.ht结尾几个文件)
2. 比对
需要用-x指定基因组索引(genome)、-U或者-1、-2输入FASTQ文件、-S输出SAM文件,最好还有日志。
hisat2 -x [index location] -U xxx.fq -S xxx.sam --summary-file xxx.align.log --new-summary
#单端 hisat2 -x [index location] -1 xxx_1.fq -2 xxx_2.fq -S xxx.sam --summary-file xxx.align.log --new-summary
#双端 比对结果解读
Aligned concordantly:两端都能合理地比对上
Aligned discordantly:两端都比对上但不合理(位置或方向等不匹配)
unpaired reads:只有一端比对上
3. 比对结果评估
reads匹配百分比
reads随机性分布(reads比对到基因上的分布均匀说明打断的随机性好)
匹配reads的GC含量和PCR偏好相关
传统基于比对-组装的方法bam
四、表达定量和差异表达分析
(一)表达水平估计
在获得转录组测序结果中的转录本及其功能注释信息后,就要根据测序reads比对到每个转录本中的数目计算该基因的表达水平,从而进行后续的分析。
表达定量方法的两大阵营
(1)Alignment-based
传统方法,以BAM文件输入
比对到基因组:Cufflinks, StringTie,结果易受测序片段长度影响
比对到转录组:eXpress, Salmon,多做一次比对耗时偏多
(2)Alignment-free
以FASTQ文件输入
quasi-mapping ≠ alignment,速度快
结果较不易受测序片段长度影响
代表工具:kallisto, Sailfish, Salmon
拓展文献:Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie, and Ballgown
Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis
1.Salmon (Quasi) 流程
salmon也可用于bam输入,此处以fasta输入为例:
(1)用salmon index(支持读取gzip)建立索引
salmon index -t transcripts.fa -i transcripts_index
#可以是fa或fa.gz文件,建立的索引文件为transcripts_index
(2)定量salmon quant分双端和单端,输入索引文件transcripts_index,输出结果文件夹transcripts_quant
#双端测序 salmon quant -i transcripts_index -l 《LIBTYPE》 -1 reads1.fq -2 reads2.fq --validateMappings -o transcripts_quant
#单端测序 salmon quant -i transcripts_index -l 《LIBTYPE》 -r reads.fq --validateMappings -o transcripts_quant
### --validateMappings 是官方推荐必加参数,先用敏感策略发现潜在mapping位点,然后打分并验证,提高准确度 注意LIBTYPE参数(1-3位字母)设置(让mapping rate正常):
(3)结果解读
输出文件夹中的quant.sf,是一个TSV文件。
#EffectiveLength:计算得到的有效长度,考虑因素包括片段长度分布和序列特异性偏差等,有些下游分析会用到 #NumReads :比对上的reads数量估计值,比对到多处的reads会根据相对丰度产生小数 #TPM (Transcripts Per Million):转录本的相对丰度估计值,可用于下游分析
原始的read counts,处理为FPKM,RPKM,TPM等……
三者区分?什么时候使用哪个指标?要看清软件输入用的指标。
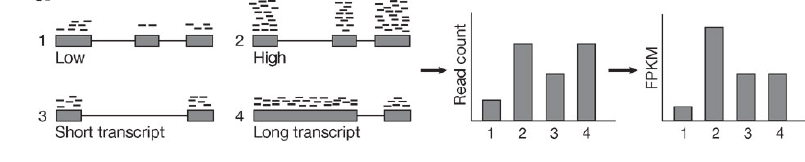
(二)差异表达分析(鉴定差异基因)
1.差异表达分析的方法和原理
需要将定量后的结果(表达矩阵)作为输入,设置好分组信息,再进行差异表达分析。
(1)方法:
基于组装:Cuffdiff, Ballgown,准确性不足
基于计数:DESeq2, edgeR(limma),前者更准确,后者支持无重复样本
→差异表达分析拓展
其他:GEO2R(针对GEO数据)
(2)标准化
RNA-Seq分析需要对基因或转录本的read counts进行normalization,因为落在一个region内的read counts取决于基因长度和测序深度。
→拓展文献Comparing the normalization methods for the differential analysis of Illumina high-throughput RNA-Seq data
2.DESeq2流程
(1)准备输入文件
①样本信息矩阵ColData:sample,condition
设计比较矩阵(contrast matrix)告诉差异分析函数应该如何对哪个因素进行比较,[默认首字母靠前的condition为对照!]
②表达矩阵countData:gene,sample,counts
如果用Salmon、Sailfish、kallisto 得到表达矩阵,那么就可以用DESeqDataSetFromTximport() 输入countData。其他导入方法还有DESeqDataSetFromMatrix()、DESeqDataSet()等

#导入salmon定量的结果 files 《- file.path(samples$run, “quant.sf”)
#files是一个个quants.sf的路径,选样本名run一列 #输入基因ID-TXNAME对应文件 tx2gene 《- read.table(file = “tx2gene.txt”, sep = “t”) #定量化生成表达矩阵 library(tximport) txi 《- tximport(files, type=“salmon”, tx2gene=tx2gene) 其中,tx2gene是转录本与基因的转换关系,可通过AnnotationHub包获取:
ah 《- AnnotationHub()
#下载数据库 sc 《- query(ah, ‘Saccharomyces cerevisiae’)
#查询物种 sc_tx 《- sc[[‘AH64985’]]
#选择ID下载详细内容 k 《- keys(sc_tx, keytype = “GENEID”)
#以基因ID为键名 df 《- select(sc_tx, keys=k, keytype = “GENEID”,columns = “TXNAME”)
#调换顺序以符合tximport要求:tx2gene 《- df[,2:1] (2)生成DESeqDataSet对象(tximport 导入为例)
library(DESeq2) dds 《- DESeqDataSetFromTximport(countData, colData = colData, design = ~ condition) #condition是数据框的因子。design说明要分析的变量
#~在R里面用于构建公式对象,~左边为因变量,右边为自变量

(3)DESeq2差异表达分析
①标准化:DESeq()
包括estimation of size factors(estimateSizeFactors), estimation of dispersion(estimateDispersons), Negative Binomial GLM fitting and Wald statistics(nbinomWaldTest)三步
dds 《- DESeq(dds)
#对dds矩阵对rawcount进行Normalize,不需事先标准化 res 《- results(dds)
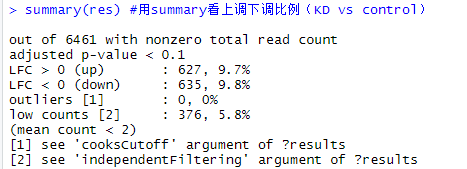
#生成结果,一个DESeqResults对象 summary(res)
#用summary看上调下调比例(默认KD vs control)、离群值等 # resOrdered 《- res[order(res$padj),]
#p值排序
②可视化:plotMA()
MA图:M表示log fold change,衡量基因表达量变化,上调还是下调。A表示每个基因的count的均值。
plotMA(res, ylim=c(-2,2))
#没有经过 statistical moderation平缓log2 fold changes的情况
library(apeglm) resLFC 《- lfcShrink(dds, coef=“condition_WT_vs_KD”, type=“apeglm”)
#经过lfcShrink 收缩log2 fold change plotMA(resLFC, ylim=c(-2,2))
③确定阈值,筛选差异表达基因
一般p-value《0.05是显著, 显著性不代表结果正确,只用于给后续的富集分析和GSEA提供排序标准和筛选。
FDR较正
假阳性随检验次数增加而增加,通常取p《0.05,1000次检验可以有50次假阳性 Bonferroni
校正:p值除以检验次数,0.05/1000=5×10-5,过于严苛导致大量假阴性 False Discovery Rate,常用
Benjamini-Hochberg 即 BH 校正方法 将一系列的p值按照从大到小排序,然后利用公式计算每个p值所对应的FDR值:FDR
= p×(n/i), p是p值,n是p值个数,最大的p值的i值为n,第二大则是n-1,依次至最小为1 将计算出来的FDR值作为新p值,如果某一个p值所对应的FDR值大于前一位p值(更大的p值)所对应的FDR值,则放弃公式计算出来的FDR值,选用与它前一位相同的值,因此会产生连续相同FDR值的现象;反之则保留计算的FDR值
返回p值对应的FDR值
res05 《- results(dds, alpha=0.05) #默认FDR小于0.1,现取阈值padj小于0.05。padj就是用BH对多重试验进行矫正 res05 summary(res05)

筛选差异显著的数据后,建立基因-FC列表,用作后续富集分析:
#提取差异表达基因集:选取上调FC》2(即log2FC》1)或下调《-2的基因 diff_gene_info 《- subset(res05, (log2FoldChange 》 1 | log2FoldChange 《 -1)) diff_genes 《- row.names(diff_gene_info)
# #提取log2FoldChange信息的列表 diff_gene_table 《- as.data.frame(diff_gene_info) geneList 《- diff_gene_table[,2]
#log2FoldChange列表用names备注对应基因名称,排序 names(geneList) = as.character(row.names(diff_gene_table)) geneList 《- sort(geneList, decreasing = TRUE) 如果只提取上调/下调,步骤也相同,总之DESeq2用于提取我们所需的基因集。
3.edgeR&limma流程
五、富集分析
富集分析在之前芯片数据分析基因的差异表达的文章中也有写到,再贴一遍富集分析介绍。
(一)GO富集分析
1.什么是GO(Gene Ontology)
基因已知的功能信息可以分为细胞组成 Cellular Component (CC)、分子功能 Molecular Function (MF)、生物过程 Biological Process (BP)三个域。
每一个域根据具体功能不同又分为不同 GO term, 有三种关系:is a,part of,regulates,通过有向无环图连接成网
通过分析一组差异基因在功能的分类关系,可以找到差异基因在那些GO分类条目富集,寻找不同样品的差异基因可能和哪些基因功能的改变有关。
官网有详细介绍和GO富集分析在线工具。
2.实现工具
在线分析工具
agriGO
利用本地数据库信息进行本地分析
R语言的clusterProfiler包,topGO包
3.GO富集分析:clusterProfiler包
(1)enrichGO()生成enrichResult对象
输入:
①待富集的基因集(如差异分析一步得到的上调基因)
不难理解这种只用了基因集的富集分析算法属于过表达分析(over representation analysis)
②物种基因数据库(OrgDb查询)
library(“clusterProfiler”) library(“org.xxx.db”)
#物种基因数据库 enrichGO_up_BP 《- enrichGO(gene = up_genes, OrgDb = “org.Sc.sgd.db”, keyType = “ENSEMBL”, ont = “BP”)
#keyType和比对GTF一致,ont三选一
(2)富集分析结果可视化
用enrichplot包实现条形图barplot()、散点图dotplot()、有向无环图plotGOgraph()的绘制:
library(“enrichplot”) barplot(enrichGO_up_BP, showCategory = 20)
#条形图 dotplot(enrichGO_up_BP, showCategory = 20)
#散点图 plotGOgraph(enrichGO_up_BP)
#有向无环图,颜色表示显著性,红色为最显著的10个 ggupset包绘制upset图对基因集合可视化:
library(“ggupset”) upsetplot(enrichGO_up_BP) #upset plot是高阶的venn图,揭示基因和基因集之间的关系
对于表达水平,可以用heatplot()绘制热图:
heatplot(enrichGO_up_BP, foldChange = gene_FC_list) #foldChange是排序后的FC-基因列表
(二)KEGG富集分析
1.什么是KEGG PATHWAY
Kyoto Encyclopedia of Genes and Genomes (KEGG)京都基因与基因组百科全书
KEGG PATHWAY: is a collection of manually drawn pathway maps representing our knowledge on the molecular interaction, reaction and relation networks for: ①Metabolism, ②Genetic Information Processing ,③Environmental Information Processing ,④Cellular Processes ,⑤Organismal Systems,⑥Human Diseases,⑦Drug Development
2.工具
(1)在线工具
KOBAS、
(2)本地工具
clusterProfiler包
3.KEGG富集分析:clusterProfiler包
还是用这个包,与GO富集分析类似做法,只不过函数是enrichKEGG(),organism走(物种缩写查询)。
enrichKEGG_up 《- enrichKEGG(gene = up_genes, organism = “sce”, keyType = ‘kegg’) barplot(enrichKEGG_up) dotplot(enrichKEGG_up)
note:著名的clusterProfiler包可以完成许多类富集分析,有空仔细研究。 →clusterProfiler包富集分析
1.数据获取
测序数据下载与处理(SRA Toolkit)
测序数据质控与过滤(fastp)
2.序列比对(SAMtools、HISAT2)
3.序列组装(StringTie、TACO)
4.表达定量和差异表达分析(Salmon、DESeq2)
5.GO和KEGG富集分析(clusterProfiler)
☆ RNA-seq方法原理
目的是要给mRNA测序,得到样本的基因表达信息。
llumina的Truseq RNA建库方法:
带Poly(T)探针的磁珠与总RNA进行杂交,吸附其中的带Poly(A)尾巴的mRNA
Mg”离子溶液处理RNA,把RNA打成短片段 被打断的mRNA片段,用随机引物逆转出第一链的cDNA,再合成双链cDNA
在双链CDNA的两端加“A“碱基,并连上”Y“型的接头
经过PCR扩增,成为可以上机的文库
起始总RNA质量控制:用电泳方法。rRNA占有总RNA的大部分,形成的峰越高/尖,RIN(RNA完整度评分值)越高,8以上质量比较好。
测到的RNA片段 mapping到基因组上,进行样品的reads在参考基因上的分布均匀性(Gene coverage)统计。两端平衡的时候表示mRNA降解少(3’高降解多)。
☆ RNA-seq的生物信息分析
一、深度测序数据获取
和EBI、DDBJ组成INSDC,数据内容相同所以找NCBI就行。
(一)NCBI常用数据库
GenBank:遗传序列数据库,收集了所有公开的DNA序列及其注释 GEO (Gene Expression Omnibus)
:收集整理各种表达芯片数据,后来加入了甲基化、lncRNA、miRNA、CNV等其他芯片,还有高通量测序数据。文献中常见GSM和GSE开头的编号,分别是GEO
Sample和GEO Series的数据 PubMed / PMC (PubMed
Central):前者把测序数据和文章联系起来,后者可以进行全文检索,无法访问校园网时可以替代Web of Knowledge
RefSeq:为所有常见生物提供非冗余、人工挑选过的参考序列,通常包含:参考基因组、参考转录组、参考蛋白序列、参考SNP信息、参考CNV信息等等
(二)测序数据的下载和处理:SRA Toolkit
测序数据序列格式
(1)FASTA:表示生物序列的文本格式,基因组和EST序列常常采用
(2)FASTQ格式:表示生物序列及其质量的文本格式
(3)ncbi SRA (Sequence Read Archive) :存储高通量测序原始数据和比对信息,把FASTQ格式文件压缩为SRA格式
绝大多数分析工具不支持SRA,需要使用配套工具包SRA Toolkit先行处理
1. SRA toolkit软件下载
在官网选择适合自己的版本下载。
#我选的ubuntu版本,其他一样,把下载链接修改一下就好了 wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.10.5/sratoolkit.2.10.5-ubuntu64.tar.gz 用conda install sra-tools失败,只好用wget方法或者手动下载到linux盘符下。把安装包下载后用tar xzvf 解压,再配置完PATH就安装好了。
检查配置:
prefetch -V 2.用SRAtoolkit下载并处理NCBI数据
将 .sra文件转换为 .fstaq.gz文件的工具。用NCBI的SRR数据测试一下。
(1)下载
理论上下载东西都可以用wget,但是太慢了。单个数据下载还好,批量下载
prefetch SRRxxxxxxx -O 。
#-O 。 指定到当前路径,否则默认路径难找
一个数据下了好久,大概1个多小时。不知道怎么优化。
(2)解压
fastq-dump SRRxxxxxxx.sra #解压后从sra文件变为fastq文件
双端测序数据要加–split-files,否则解压后两端的数据不会分开,难以被其他软件读取 如果所用分析软件支持读取gzip,建议加上–gzip,将解压后的数据用gzip压缩,避免占用过多空间
fastq-dump --split-files --gzip xxx.sra
(三)测序数据质控与过滤: fastp
输出HTML和JSON报告,前者方便阅读,后者方便软件读取
单端:fastp -i raw.fq -o clean.fq
双端:fastp -i raw_1.fq -I raw_2.fq -o clean_1.fq -O clean_2.fq
有必要附加的参数:-l 36 -j xxx.json -h xxx.html
默认报告文件名 fastp.json 和 fastp.html,处理多个样本时极易互相覆盖,建议改为样本名称
fastp参数设置
# I/O options 输入输出序列文件 -i 《单端-输入文件名》 -o 《单端-输出文件名》 -I 《双端-输入文件名》 -O 《双端-输出文件名》
#过滤后的最短序列长度 -l 36 #默认15,建议设为36或40 # reporting options 报告参数 -j 《the json format report file name 》 -h 《the html format report file name 》 -R “report_title”
二、序列比对:HISAT2
注释格式介绍
(1)GFF/GTF格式:一般用于基因组和基因注释
(2)SAM格式:储存序列比对到基因组上的信息的文本格式,
(3)BAMS:SAM的基础上,用二进制 (Binary) 编码,以便压缩体积。
压缩率高于gzip,绝大多数下游分析工具使用
(4)CRAM:在BAM的基础上,借助参考序列,进一步减少空间占用
用SAMtools将SAM转化为BAM或CRAM格式
samtools sort -o xxx.bam xxx.sam samtools sort -o xxx.cram --reference ref.fa -O cram xxx.sam #加-O指定输出格式 建立索引以便快速读取
samtools index xxx.bam samtools index xxx.cram
为什么要比对 (align / map)
locate:测序所得的短序列在基因组的哪个位置
variant:如果个别碱基与基因组不一致,是测序错误还是变异
比对软件工作过程
根据基因组序列FASTA和注释GTF,通过一定的算法编制索引
FASTQ比对到索引,生成SAM文件
如HISAT 和 Bowtie 基于BWT算法。
1. 用HISAT2建立索引
有注释:基因组GTF文件Splice Sites和Exons信息,与基因组序列一起用于建立索引
hisat2_extract_splice_sites.py genes.gtf 》 splicesites.txt hisat2_extract_exons.py genes.gtf 》 exons.txt hisat2-build --ss splicesites.txt --exon exons.txt genome.fa genome 没注释:直接用基因组序列建立索引
hisat2-build genome.fa genome 结果产生索引文件genome(指向.ht结尾几个文件)
2. 比对
需要用-x指定基因组索引(genome)、-U或者-1、-2输入FASTQ文件、-S输出SAM文件,最好还有日志。
hisat2 -x [index location] -U xxx.fq -S xxx.sam --summary-file xxx.align.log --new-summary
#单端 hisat2 -x [index location] -1 xxx_1.fq -2 xxx_2.fq -S xxx.sam --summary-file xxx.align.log --new-summary
#双端 比对结果解读
Aligned concordantly:两端都能合理地比对上
Aligned discordantly:两端都比对上但不合理(位置或方向等不匹配)
unpaired reads:只有一端比对上
3. 比对结果评估
reads匹配百分比
reads随机性分布(reads比对到基因上的分布均匀说明打断的随机性好)
匹配reads的GC含量和PCR偏好相关
传统基于比对-组装的方法bam
四、表达定量和差异表达分析
(一)表达水平估计
在获得转录组测序结果中的转录本及其功能注释信息后,就要根据测序reads比对到每个转录本中的数目计算该基因的表达水平,从而进行后续的分析。
表达定量方法的两大阵营
(1)Alignment-based
传统方法,以BAM文件输入
比对到基因组:Cufflinks, StringTie,结果易受测序片段长度影响
比对到转录组:eXpress, Salmon,多做一次比对耗时偏多
(2)Alignment-free
以FASTQ文件输入
quasi-mapping ≠ alignment,速度快
结果较不易受测序片段长度影响
代表工具:kallisto, Sailfish, Salmon
拓展文献:Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie, and Ballgown
Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis
1.Salmon (Quasi) 流程
salmon也可用于bam输入,此处以fasta输入为例:
(1)用salmon index(支持读取gzip)建立索引
salmon index -t transcripts.fa -i transcripts_index
#可以是fa或fa.gz文件,建立的索引文件为transcripts_index
(2)定量salmon quant分双端和单端,输入索引文件transcripts_index,输出结果文件夹transcripts_quant
#双端测序 salmon quant -i transcripts_index -l 《LIBTYPE》 -1 reads1.fq -2 reads2.fq --validateMappings -o transcripts_quant
#单端测序 salmon quant -i transcripts_index -l 《LIBTYPE》 -r reads.fq --validateMappings -o transcripts_quant
### --validateMappings 是官方推荐必加参数,先用敏感策略发现潜在mapping位点,然后打分并验证,提高准确度 注意LIBTYPE参数(1-3位字母)设置(让mapping rate正常):
(3)结果解读
输出文件夹中的quant.sf,是一个TSV文件。
#EffectiveLength:计算得到的有效长度,考虑因素包括片段长度分布和序列特异性偏差等,有些下游分析会用到 #NumReads :比对上的reads数量估计值,比对到多处的reads会根据相对丰度产生小数 #TPM (Transcripts Per Million):转录本的相对丰度估计值,可用于下游分析
原始的read counts,处理为FPKM,RPKM,TPM等……
三者区分?什么时候使用哪个指标?要看清软件输入用的指标。
(二)差异表达分析(鉴定差异基因)
1.差异表达分析的方法和原理
需要将定量后的结果(表达矩阵)作为输入,设置好分组信息,再进行差异表达分析。
(1)方法:
基于组装:Cuffdiff, Ballgown,准确性不足
基于计数:DESeq2, edgeR(limma),前者更准确,后者支持无重复样本
→差异表达分析拓展
其他:GEO2R(针对GEO数据)
(2)标准化
RNA-Seq分析需要对基因或转录本的read counts进行normalization,因为落在一个region内的read counts取决于基因长度和测序深度。
→拓展文献Comparing the normalization methods for the differential analysis of Illumina high-throughput RNA-Seq data
2.DESeq2流程
(1)准备输入文件
①样本信息矩阵ColData:sample,condition
设计比较矩阵(contrast matrix)告诉差异分析函数应该如何对哪个因素进行比较,[默认首字母靠前的condition为对照!]
②表达矩阵countData:gene,sample,counts
如果用Salmon、Sailfish、kallisto 得到表达矩阵,那么就可以用DESeqDataSetFromTximport() 输入countData。其他导入方法还有DESeqDataSetFromMatrix()、DESeqDataSet()等
#导入salmon定量的结果 files 《- file.path(samples$run, “quant.sf”)
#files是一个个quants.sf的路径,选样本名run一列 #输入基因ID-TXNAME对应文件 tx2gene 《- read.table(file = “tx2gene.txt”, sep = “t”) #定量化生成表达矩阵 library(tximport) txi 《- tximport(files, type=“salmon”, tx2gene=tx2gene) 其中,tx2gene是转录本与基因的转换关系,可通过AnnotationHub包获取:
ah 《- AnnotationHub()
#下载数据库 sc 《- query(ah, ‘Saccharomyces cerevisiae’)
#查询物种 sc_tx 《- sc[[‘AH64985’]]
#选择ID下载详细内容 k 《- keys(sc_tx, keytype = “GENEID”)
#以基因ID为键名 df 《- select(sc_tx, keys=k, keytype = “GENEID”,columns = “TXNAME”)
#调换顺序以符合tximport要求:tx2gene 《- df[,2:1] (2)生成DESeqDataSet对象(tximport 导入为例)
library(DESeq2) dds 《- DESeqDataSetFromTximport(countData, colData = colData, design = ~ condition) #condition是数据框的因子。design说明要分析的变量
#~在R里面用于构建公式对象,~左边为因变量,右边为自变量
(3)DESeq2差异表达分析
①标准化:DESeq()
包括estimation of size factors(estimateSizeFactors), estimation of dispersion(estimateDispersons), Negative Binomial GLM fitting and Wald statistics(nbinomWaldTest)三步
dds 《- DESeq(dds)
#对dds矩阵对rawcount进行Normalize,不需事先标准化 res 《- results(dds)
#生成结果,一个DESeqResults对象 summary(res)
#用summary看上调下调比例(默认KD vs control)、离群值等 # resOrdered 《- res[order(res$padj),]
#p值排序
②可视化:plotMA()
MA图:M表示log fold change,衡量基因表达量变化,上调还是下调。A表示每个基因的count的均值。
plotMA(res, ylim=c(-2,2))
#没有经过 statistical moderation平缓log2 fold changes的情况
library(apeglm) resLFC 《- lfcShrink(dds, coef=“condition_WT_vs_KD”, type=“apeglm”)
#经过lfcShrink 收缩log2 fold change plotMA(resLFC, ylim=c(-2,2))
③确定阈值,筛选差异表达基因
一般p-value《0.05是显著, 显著性不代表结果正确,只用于给后续的富集分析和GSEA提供排序标准和筛选。
FDR较正
假阳性随检验次数增加而增加,通常取p《0.05,1000次检验可以有50次假阳性 Bonferroni
校正:p值除以检验次数,0.05/1000=5×10-5,过于严苛导致大量假阴性 False Discovery Rate,常用
Benjamini-Hochberg 即 BH 校正方法 将一系列的p值按照从大到小排序,然后利用公式计算每个p值所对应的FDR值:FDR
= p×(n/i), p是p值,n是p值个数,最大的p值的i值为n,第二大则是n-1,依次至最小为1 将计算出来的FDR值作为新p值,如果某一个p值所对应的FDR值大于前一位p值(更大的p值)所对应的FDR值,则放弃公式计算出来的FDR值,选用与它前一位相同的值,因此会产生连续相同FDR值的现象;反之则保留计算的FDR值
返回p值对应的FDR值
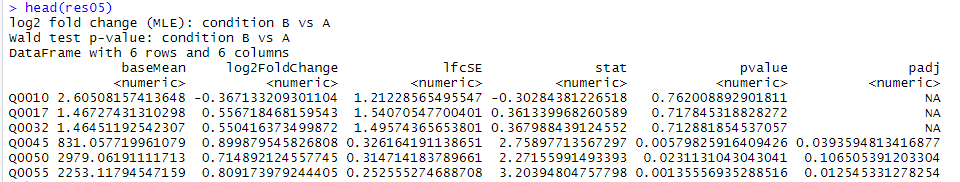
res05 《- results(dds, alpha=0.05) #默认FDR小于0.1,现取阈值padj小于0.05。padj就是用BH对多重试验进行矫正 res05 summary(res05)
筛选差异显著的数据后,建立基因-FC列表,用作后续富集分析:
#提取差异表达基因集:选取上调FC》2(即log2FC》1)或下调《-2的基因 diff_gene_info 《- subset(res05, (log2FoldChange 》 1 | log2FoldChange 《 -1)) diff_genes 《- row.names(diff_gene_info)
# #提取log2FoldChange信息的列表 diff_gene_table 《- as.data.frame(diff_gene_info) geneList 《- diff_gene_table[,2]
#log2FoldChange列表用names备注对应基因名称,排序 names(geneList) = as.character(row.names(diff_gene_table)) geneList 《- sort(geneList, decreasing = TRUE) 如果只提取上调/下调,步骤也相同,总之DESeq2用于提取我们所需的基因集。
3.edgeR&limma流程
五、富集分析
富集分析在之前芯片数据分析基因的差异表达的文章中也有写到,再贴一遍富集分析介绍。
(一)GO富集分析
1.什么是GO(Gene Ontology)
基因已知的功能信息可以分为细胞组成 Cellular Component (CC)、分子功能 Molecular Function (MF)、生物过程 Biological Process (BP)三个域。
每一个域根据具体功能不同又分为不同 GO term, 有三种关系:is a,part of,regulates,通过有向无环图连接成网
通过分析一组差异基因在功能的分类关系,可以找到差异基因在那些GO分类条目富集,寻找不同样品的差异基因可能和哪些基因功能的改变有关。
官网有详细介绍和GO富集分析在线工具。
2.实现工具
在线分析工具
agriGO
利用本地数据库信息进行本地分析
R语言的clusterProfiler包,topGO包
3.GO富集分析:clusterProfiler包
(1)enrichGO()生成enrichResult对象
输入:
①待富集的基因集(如差异分析一步得到的上调基因)
不难理解这种只用了基因集的富集分析算法属于过表达分析(over representation analysis)
②物种基因数据库(OrgDb查询)
library(“clusterProfiler”) library(“org.xxx.db”)
#物种基因数据库 enrichGO_up_BP 《- enrichGO(gene = up_genes, OrgDb = “org.Sc.sgd.db”, keyType = “ENSEMBL”, ont = “BP”)
#keyType和比对GTF一致,ont三选一
(2)富集分析结果可视化
用enrichplot包实现条形图barplot()、散点图dotplot()、有向无环图plotGOgraph()的绘制:
library(“enrichplot”) barplot(enrichGO_up_BP, showCategory = 20)
#条形图 dotplot(enrichGO_up_BP, showCategory = 20)
#散点图 plotGOgraph(enrichGO_up_BP)
#有向无环图,颜色表示显著性,红色为最显著的10个 ggupset包绘制upset图对基因集合可视化:
library(“ggupset”) upsetplot(enrichGO_up_BP) #upset plot是高阶的venn图,揭示基因和基因集之间的关系
对于表达水平,可以用heatplot()绘制热图:
heatplot(enrichGO_up_BP, foldChange = gene_FC_list) #foldChange是排序后的FC-基因列表
(二)KEGG富集分析
1.什么是KEGG PATHWAY
Kyoto Encyclopedia of Genes and Genomes (KEGG)京都基因与基因组百科全书
KEGG PATHWAY: is a collection of manually drawn pathway maps representing our knowledge on the molecular interaction, reaction and relation networks for: ①Metabolism, ②Genetic Information Processing ,③Environmental Information Processing ,④Cellular Processes ,⑤Organismal Systems,⑥Human Diseases,⑦Drug Development
2.工具
(1)在线工具
KOBAS、
(2)本地工具
clusterProfiler包
3.KEGG富集分析:clusterProfiler包
还是用这个包,与GO富集分析类似做法,只不过函数是enrichKEGG(),organism走(物种缩写查询)。
enrichKEGG_up 《- enrichKEGG(gene = up_genes, organism = “sce”, keyType = ‘kegg’) barplot(enrichKEGG_up) dotplot(enrichKEGG_up)
note:著名的clusterProfiler包可以完成许多类富集分析,有空仔细研究。 →clusterProfiler包富集分析

 举报
举报




